
Episode 094: Heme Consult Series: Cold Agglutinin Disease
In this week’s episode, we finish our series on hemolytic anemias with a discussion on cold agglutinin disease along with a few other causes of acquired hemolytic anemia.
In our intro, Vivek also shed light on the drama surrounding the recent season of the Golden Bachelor. Wanna know more? Click here: Drama for the Golden Bachelor. (WARNING: spoiler alert)
What is cold agglutinin disease?
Cold agglutinin disease is a rare type of acquired auto-immune hemolytic anemia
Similar to warm AIHA, cold agglutinin disease results from a pathogenic autoantibody that targets an antigen that is found on normal red blood cells
Unlike in wAIHA (where an IgG antibody causes hemolysis), the offending antibody in cold agglutinin disease is an IgM antibody. Remember that IgM is larger than IgG and forms pentamers in the blood.
These offending IgM pentamers (ie, the “cold agglutinins”) target epitopes on the surface of the RBCs (either Big “I” or little “i”) that are universally present on all RBCs
Because of their size, each IgM pentamer can bind multiple RBCs together causing visible agglutination
Once the IgM pentamers bind to RBCs, they can then trigger activation of the classical complement system. This eventually leads to the RBC being coated in C3b.
This C3b coating is what allows cells to be recognized by macrophages in the reticuloendothelial system (within the liver and spleen), and ultimately leads to sequestration/destruction of the RBCs. This is why the hemolysis in cold agglutinin disease is primarily considered an extravascular hemolysis.
A portion of the RBCs will survive and have C3b cleaved further, leaving behind C3d on the surface of the RBC. C3d is able to be detected on a DAT, so this is why the classic finding for cold agglutinin disease on a DAT is IgG negative and C3 positive.
While the majority of hemolysis is extravascular and due to macrophage-clearance of C3b coated RBCs, there may be a portion of cells which undergo further complement activation. In the most severe cases of cold agglutinin disease, further complement activation leads to the assembly of the membrane attack complex (MAC) on the RBC surface which causes intravascular hemolysis.
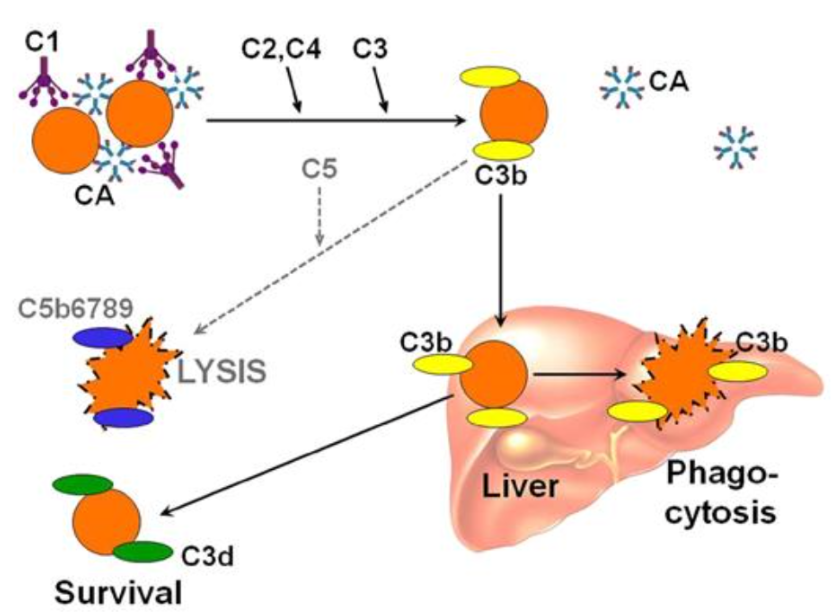
Image source: Sigbjørn Berentsen; Cold agglutinin disease. Hematology Am Soc Hematol Educ Program 2016; 2016 (1): 226–231.
Why do we call it “cold”?
At normal body temperature, these antibodies typically do NOT bind to RBCs
For this reason, it is important to keep blood samples at normal body temperature after drawn or they may agglutinate within the tube before they can be tested in the laboratory
These “cold agglutinins” have the highest binding activity when below normal body temperature (with optimal binding occurring around 4° C).
Although optimal binding occurs well below body temperature, these antibodies can still bind to some degree at higher temperatures
The temperature range in which an antibody is able to significantly bind RBCs can vary between patients. The highest temperature at which the antibody causes binding is called the thermal amplitude
In general, the higher the thermal amplitude = the higher the pathogenicity of the antibody (ie, the lower the thermal amplitude, the colder the blood has to be for the antibody to become problematic and vice versa)
Clinically relevant cold agglutinin antibodies typically have a thermal amplitude of 28-30° C or higher. Antibodies with a lower thermal amplitude may be present but non-pathogenic (ie, they only bind at temperatures far below what the blood will ever actually experience)
This temperature-dependent binding is why areas that are lower than core body temperature (ex: fingers, toes, nose, etc.) are more likely to be symptomatic when someone is in a cold external environment
How do we diagnose cold agglutinin disease?
In order to diagnose cold agglutinin disease, we need to prove that there is an antibody present that has caused the complement system to activate against RBCs and that this antibody is temperature-dependent.
To prove this, we need to check a DAT as well as measure antibody titers at a cold temperature (4°C)
There are two types of DATs that you may see reported: polyspecific and monospecific
Polyspecific DATs will react if either C3 or IgG are present on RBCs
Monospecific DATs test for these two things separately, allowing you to determine exactly which one (or both) is present. In the case of cold agglutinin disease, we want to determine whether C3d is present on RBCs as this only happens if the complement system has been inappropriately targeting RBCs.
Remember that the titer of an antibody refers to how much you can dilute the specimen and still detect the offending antibody
Higher titers = the sample can be diluted more times and you can still detect this antibody, meaning it was present in higher amounts in the original sample.
Putting this together, there are two requirements to make a diagnosis of cold agglutinin disease in a patient presenting with a hemolytic anemia:
Monospecific DAT positive for C3d and negative (or weakly positive) for IgG
Cold agglutinin (antibody) titer greater than 1:64 when measured at a temperature of 4°C
What causes cold agglutinin disease to occur?
“Cold agglutinin disease” is a term used when this condition occurs without some other obvious precipitating factor.
This is usually associated with some undiagnosed lymphoproliferative disorder involving an abnormal B-cell clone that produces these pathogenic antibodies
“Cold agglutinin syndrome” is a term used when this phenomenon occurs secondary to another precipitating condition (such as overt malignancy, infection, or an another underlying autoimmune condition)
Examples of precipitating malignancies include indolent blood cancers such as marginal zone lymphoma, CLL, or lymphoplasmacytic lymphoma (formerly known as Waldenstrom’s macroglobulinemia)
Unlike in cold agglutinin disease, these conditions are more obviously present based on other clinical/radiologic findings.
What types of testing should we send to evaluate for a lymphoproliferative condition?
Flow cytometry from peripheral blood - this can help evaluate for a clonal B-cell population.
Even if a clonal B-cell population is identified, it can still be considered cold agglutinin disease as long as this population does not meet criteria for a formal diagnosis of another condition like CLL (in which case it would be considered to be a secondary process, aka cold agglutinin syndrome)
SPEP and serum free light chains - >90% of patients will have an M-spike positive for IgM kappa.
Bone marrow biopsy - while this is not emergent, it should be considered in the outpatient setting to help evaluate for an underlying occult hematologic malignancy
How do you manage cold agglutinins disease?
In managing cold agglutinin disease, establishing a foundation of non-pharmacological management is pivotal.
Temperature management: Avoiding cold exposure in acral areas, particularly the head, feet, and hands.
Blood transfusions: Use a blood warmer to preemptively mitigate unnecessary hemolysis
Seasonal relocation: Relocating to warmer areas during winter seasons to minimize impacts of cold related triggers.
Surgical preparedness: Pre-surgical planning is essential to lessen brisk hemolysis intraoperatively
Steroids are typically not beneficial in cold agglutinins disease, only resulting in remission in <20% of patients.
Splenectomy is not beneficial as the hemolysis is complement mediated and can also occur in the liver.
In this small study by Berentsen, no patients with cold agglutinins disease obtained CR with splenectomy.
Asymptomatic or mildly symptomatic individuals can be managed with non-pharmacological management, as more aggressive treatments have not been shown to be of benefit .
If patients are symptomatic, what treatments should we consider?

Image source: Berenstein. Blood (2021) 137 (10): 1295–1303.
Navigating the treatment landscape for cold agglutinin disease is depicted in “How I Treat Cold Agglutinin Disease” concept map.
Factors to consider:
Patient’s fitness level
Presence of symptoms
Individual responses to interventions with strategies
Remember, there will often be a monoclonal B cell population driving this process.
Despite the absence of randomized clinical trials for CAD, there are many retrospective and prospective studies supporting the efficacy of B-cell targeted therapy to reduce the burden of disease.
Table 2 in “How I Treat” provides a comprehensive overview of the various medications with their associated response rates.
Rituximab is used as first line therapy.
It is generally well-tolerated and can be given as monotherapy during the winter months, while monitoring titers as you progress through therapy.
There is no data for maintenance rituximab therapy.
For robust and fit patients without comorbidities, and with a confirmed cell line driving the cold agglutinins disease, you could consider starting bendamustine-rituximab as first line therapy.
Consider bone marrow biopsy to attempt to uncover an underlying process is warranted at this juncture.
Firstly, it serves as a diagnostic tool to uncover an indolent primary B-cell malignancy.
Secondly, it could open up more targeted therapeutic options to definitively treat the cold agglutinins disease.
Other second line options include ibrutinib, bortezomib, rituximab-fludarabine.
Since this disease is complement driven, is complement inhibition an option?
In 2022, a complement inhibitor, known as sutimlimab was approved as the result of two large studies known as CARDINAL and CADENZA.
Sutimlimab acts by blocking the C1s, subsequently inhibiting the classical completement cascade.
Unfortunately, this medication only acts as a temporary bandage in patients with cold aggultins disease, as the monoclonal B-clone is still at play, so definitive therapies should be considered.

Image source: Figure 3. Berenstein. Blood (2019) 133:885-886.
Other causes of acquired hemolytic anemias.
Paroxysmal nocturnal hemoglobinuria:
An acquired genetic defect that leads to loss of surface CD55 and 59 expression, which in turn leads to complement-mediated hemolysis (We will discuss this more in a future episode, as this disorder has implications in a variety of diseases)
Spur cell anemia:
Most commonly in patients with liver disease is spur cell anemia.
Spur cells or acanthocytes which will have irregular projections from the cell.
This can cause profound hemolysis in patients with advanced liver disease and unfortunately without fixing the liver, supportive care is what is best.
The presence of spur cell anemia has been correlated with increased rates of mortality
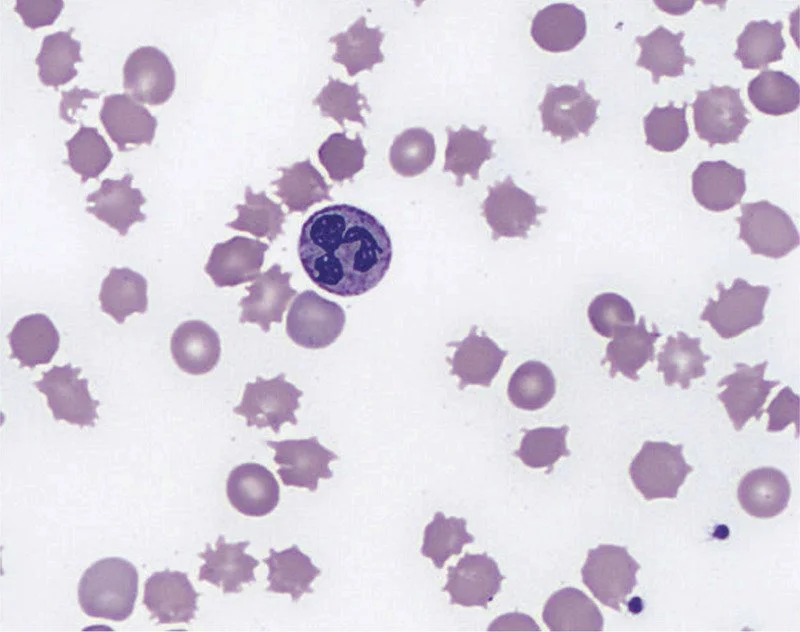
Image source: Keyserling and Koprowski. NEJM. (2019) 379:774
Medication-induced hemolysis:
This highlights why doing a good medication reconciliation is so important.
Notorious drugs include many antibiotics, including penicillins and cephalosporins, sulfonamides, and tetracyclines.
Briefly, these drugs can cause hemolysis in one of three ways:
Either they act as a hapten and coat the RBC surface, thereby resulting in complement activation and/or phagocytosis
The drug can bind a carrier protein and trigger antibody formation. If you can imagine, how this carrier protein attached to the drug, which is then attached to antibodies then binds the RBC via its normal receptors. This bulky complex can then result in complement activation.
Lastly, some drugs induce the development of autoimmune antibodies that directly interact with the RBC surface and result in phagocytosis
Important reminder: Keep an open mind and a broad differential when seeing a patient for hemolysis. More often than not, underlying pathologies or iatrogenic causes should be investigated, while also ruling out AIHA.
References:
Wintrobe’s Hematology, 14th Edition
https://ashpublications.org/blood/article/137/10/1295/475028/How-I-treat-cold-agglutinin-disease : How I treat article for CAD
https://www.nejm.org/doi/full/10.1056/NEJMoa2027760: CARDINAL study leading to approval of sutimlimab
https://ashpublications.org/blood/article/140/9/980/485526/Sutimlimab-in-patients-with-cold-agglutinin : CADENZA study leading to approval of sutimlimab
https://pubmed.ncbi.nlm.nih.gov/24325340: Poor prognosis with Spur Cell Anemia
The crew behind the magic:
Show outline: Ronak Mistry
Production and hosts: Ronak Mistry, Vivek Patel, Dan Hausrath
Editing: Resonate Recordings
Shownotes: Megan Connor, Sean Taasan
Social media management: Ronak Mistry

We are proud to partner with HemOnc.org!
Want to learn more about the trials that lead to the regimens discussed today? What about dosing schedules? See links in the show notes for a link to HemOnc.org