
Episode 092: Heme Consult Series: Congenital Causes of Hemolytic Anemia
In the next part of this heme consult series, we discuss several congenital causes of hemolytic anemias. These diseases are relatively rare, but in patients presenting with concerns for hemolysis on history and on labs, but with a negative DAT, it is important to have these in your differential diagnosis! We take you through how to think about these disorders, their diagnosis, and management.
If you have not done so already, be sure to check out Episode 091 where we discuss our initial approach to the diagnosis of hemolytic anemias. We also discuss the most common inherited cause of hemolytic anemia, G6PD deficiency, in that episode!
When should we suspect congenital causes of hemolytic anemia?
Suspect in patients who present with labs consistent with hemolytic anemia (acute drop in hemoglobin, elevated LDH, elevated reticulocyte counts, low haptoglobin, elevated indirect bilirubin), but with negative DAT
When it comes to inherited hemolytic anemias, how can we put these into buckets to easily remember?
Hemoglobinopathies: Sickle cell disease, thalassemias (more about these in later series)
Membrane disorders: Either a structural issue or an issue with maintaining the hydration of the cell
Enzyme disorders
What are common membranopathies that we should consider?
Very important to look at peripheral smear to use as the guide
Hereditary spherocytosis:
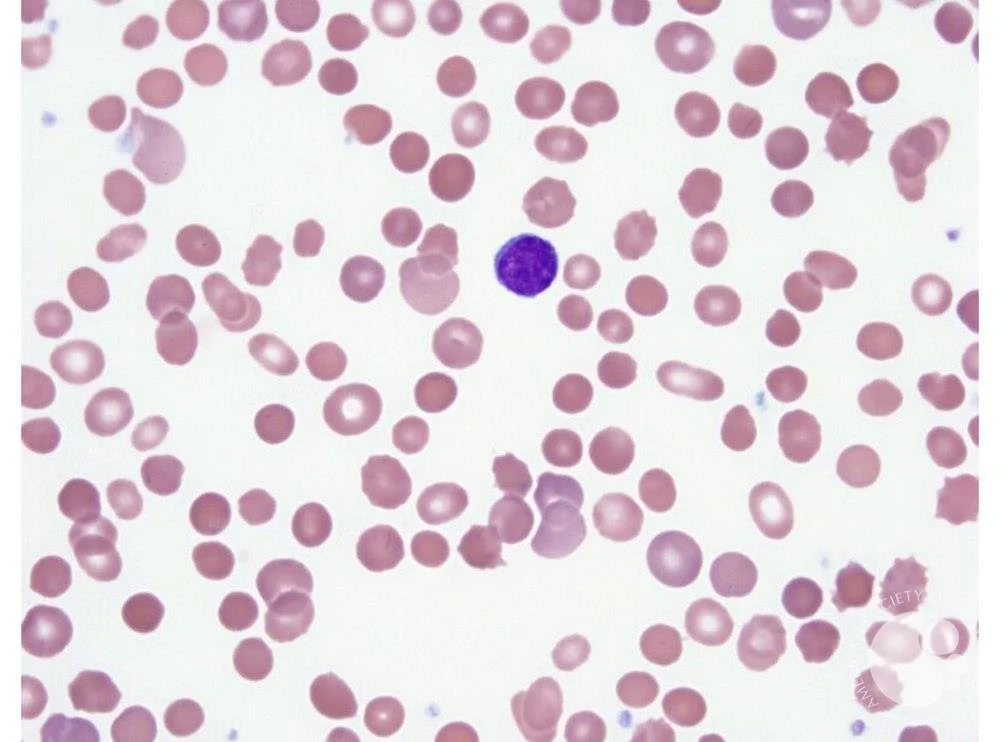
Image Source: ASH Image Bank- Hereditary spherocytosis (https://imagebank.hematology.org/image/60308/spherocytes--hereditary-spherocytosis) . No Copyright infringement intended
Epidemiology: Most common in people of Northern European descent
Inheritance:
Autosomal dominant manner (most often)
However about 25% will have non-dominant inheritance (due to de novo mutations)
Evaluation:
Smear with spherocytes
Underlying problem:
Problem with VERTICAL interactions (“sph-VERO-cytosis”)
The defective proteins are those involved in anchoring the membrane to the inner scaffolding of the RBC:
The alpha and beta spectrin molecules that look like twizzlers and provide the inner scaffold
Ankyrin which connects that alpha-beta spectrin to the membrane proteins
Proteins Band 3 and Protein 4.2, which also play a role in membrane stability
Why hemolysis?
Because they have a healthy spleen! The mis-shapen RBCs then get eaten up by the macrophages
There is also something called “conditioning” where the splenic environment accelerates membrane loss and spherocyte formation, which also makes the RBCs prone to hemolysis.
How to make the diagnosis:
Anemia, reticulocytosis, increased mean corpuscular hemoglobin concentration (due to loss of surface area; >/= 35), increased RDW, normal MCV, hyperbilirubinemia
Positive “osmotic fragility test” - the basic idea here is that the cells are put into more hypotonic solution, meaning water enters the cell. Since they have decreased membrane surface area, it takes less water to cause the cells to break.
Another diagnostic test is called the eosin-5-maleimide (EMA), which uses flow cytometry to make the diagnosis.
Molecular studies can also be performed
How do we treat it?
HS is graded mild to severe
Treatment is going to be dictated by the severity of their disease.
All patients need folic acid
Splenectomy used to be indicated for everyone, but we now understand that this is a high risk procedure also associated with increased risk of infection, so this is reserved for patients with worse disease.
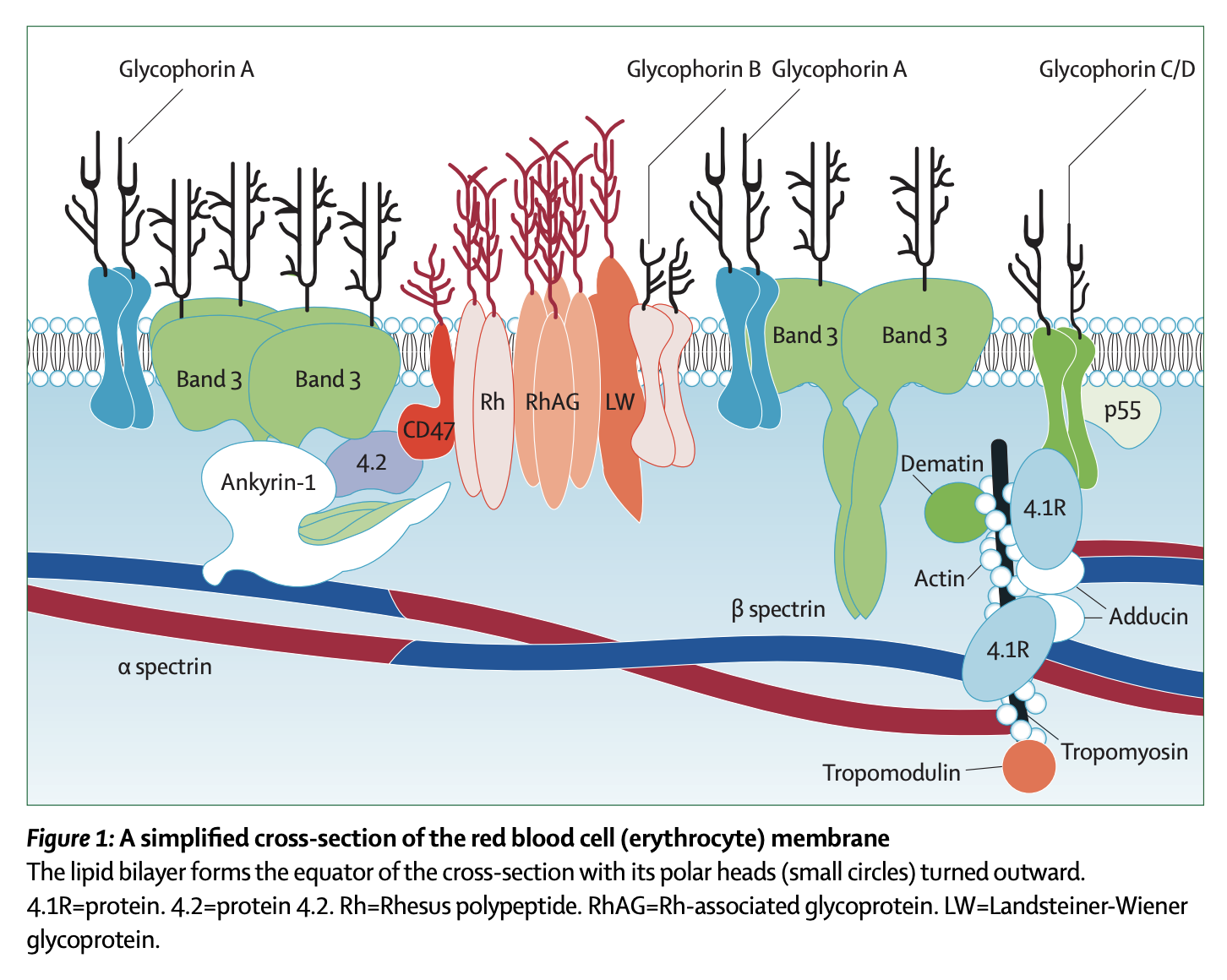
Image source: Figure 1. Silverio Perrotta, Patrick G Gallagher, Narla Mohandas. “Hereditary Spherocytosis,” Lancet 2008. No copyright infringement intended
Hereditary elliptocytosis
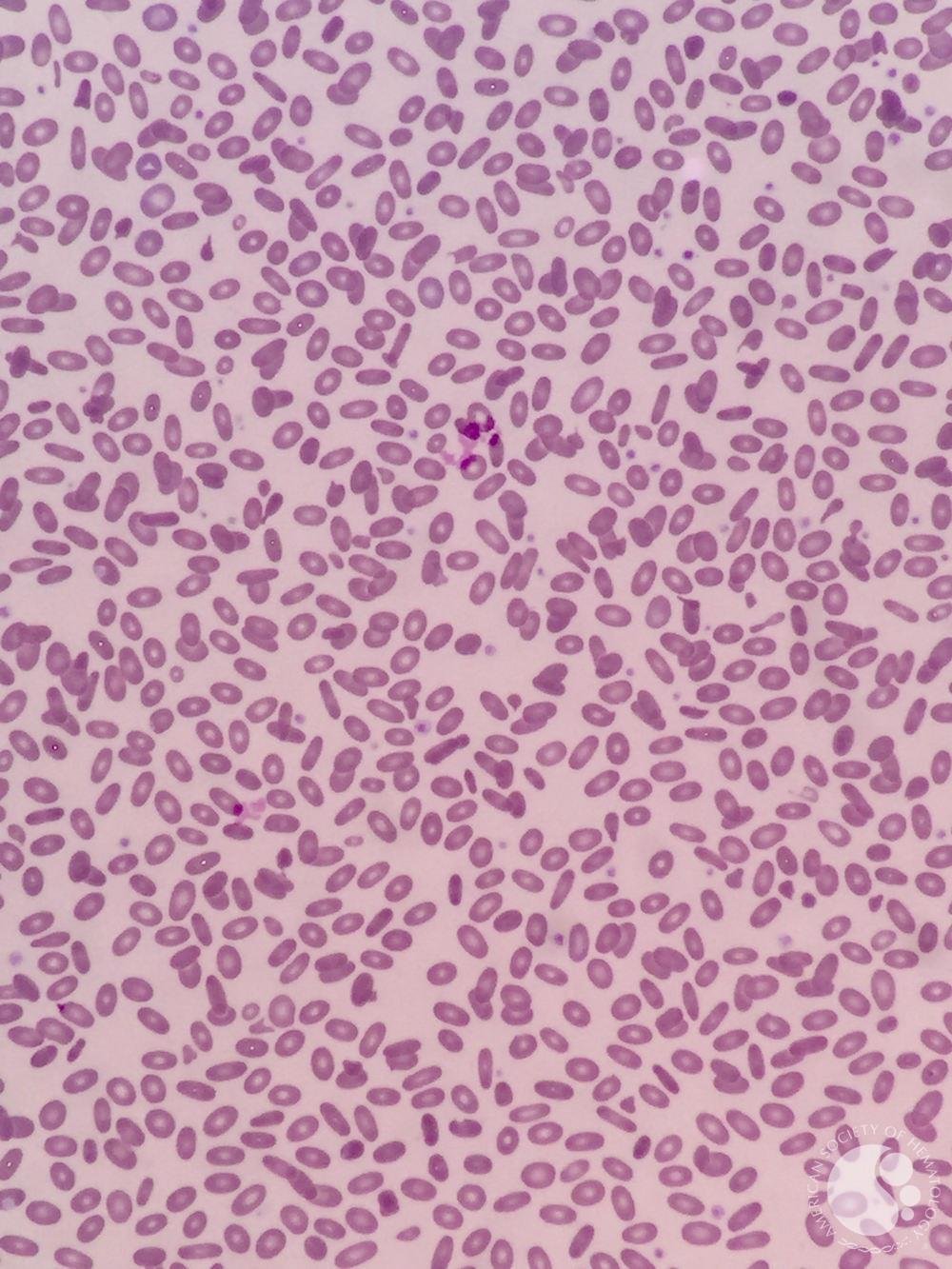
Image source: ASH Image Bank - Hereditary elliptocytosis (https://imagebank.hematology.org/image/61156/hereditary-elliptocytosis). No copyright infringement intended.
Epidemiology: More common in African and Mediterranean descent
Inheritance: Autosomal dominant with rare de novo mutations
Evaluation: Smear with elliptocytes
Subtypes:
Common HE: Patients are often asymptomatic or symptomatic when they are sick and essentially their abnormal cells are found incidentally
Hemolytic HE: Usually moderate to severely symptomatic; these patients will often have an enlarged spleen.
Hereditary pyropoikilocytosis: looks like thermal burns, and basically creates a moderate to severe hemolytic anemia.
Underlying problem:
Problem with HORIZONTAL interaction proteins
Interestingly this is also in alpha and beta-spectrin, but these are going to be different than the HS mutation; the HE mutations prevent the formation of the alpha-beta spectrin tetramer.
There are rarely other causes, as well.
Why hemolysis: Same as HS - a normal spleen
How to make the diagnosis:
Anemia with elevated LDH and bilirubin
In hemolytic HE and hereditary pyropoikilocytosis, the MCV will be really small
The osmotic fragility test will be normal in non-hemolytic HE, but increased in other types.
There are genetic tests that can also confirm the diagnosis
How do we treat it?
Treatment is again going to be dictated by the severity of the disease
All patients should be on folic acid
PRN transfusions may be needed
Splenectomy can be considered in severe causes
Stomatocytosis
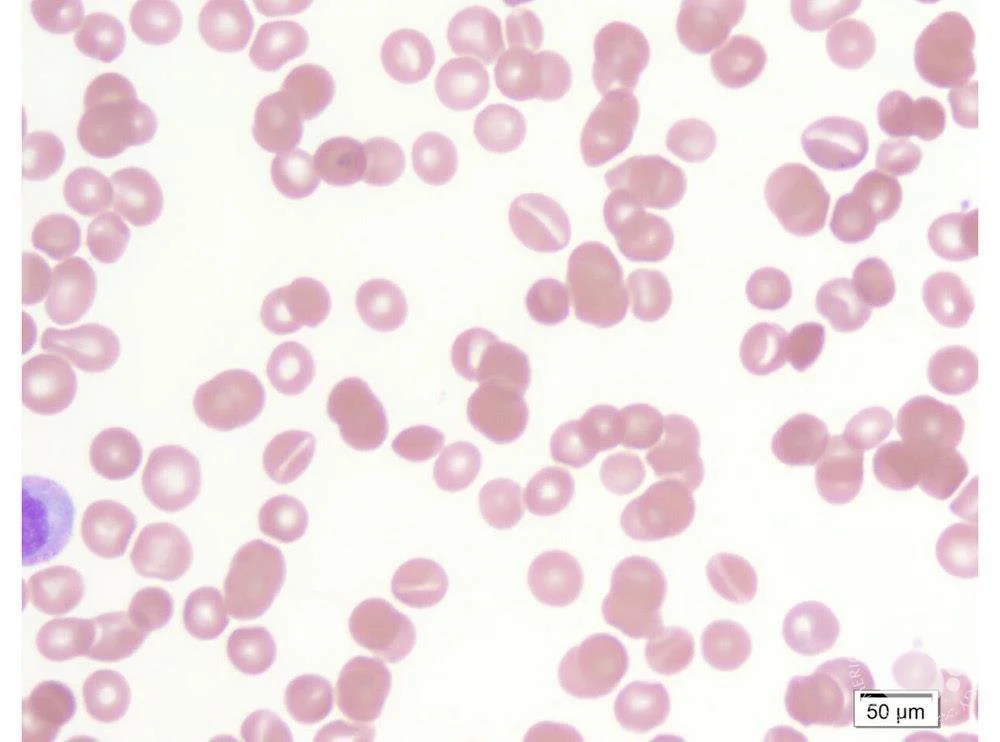
Image source: ASH Image Bank - Stomatocytes (https://imagebank.hematology.org/image/60309/stomatocytes). No copyright infrigement intended.
When to suspect: abundance of coffee-bean or “mouth-shaped” cells due to abnormal Na/K pumps on the cell surface
Two outcomes:
Hydrocytosis: cells are overhydrated
Cells are big
Autosomal dominant inheritance
Elevated MCV (110-15) and low MCHC (more water inside so hean concentration of hemoglobin is diluted)
Xerocytosis: Cells have less water
Cells are small!
Autosomal dominant inheritance
Most common mutation is in PIEZO-1!
Low MCV (small cells) with high MCHC (more concentrated hemoglobin)
Important to remember that high rates of thrombosis after splenectomy in these patients (Why? Hot off the Press in Blood 2024: https://ashpublications.org/blood/article/143/4/357/506380/Deciphering-and-disrupting-PIEZO1-TMEM16F)
What are examples of enzyme deficiencies that we should know about?
There is a very long list of possible enzyme deficiencies.
In Episode 091, we discussed G6PD deficiency already
Another important one to know is PK Deficiency
PK Deficiency
Epidemiology: 1 per 20,000 Caucasians
of all of the glycolytic and nucleotide enzymopathies, this is the most common and the most studied.
Inheritance: Autosomal recessive disorder.
Over 300 mutations!
Most patients are compound heterozygotes; rarely homozygous
Patients with one abnormal PK copy are normal
Clinical presentation: Variable (hydrops fetalis, neonatal jaundice, variable presentation in adults)
Diagnosis:
Characterized by anemia, which can be variable.
Peripheral smear will often have cells of varying shapes and sizes
There is often a high reticulocyte count (since retics have normal mitochondria so they can bypass the need for PK!)
PK enzyme activity levels should be checked if this is suspected, but leukocytes must be reduced since they have PK isomers that are not affected by this mutation.
Treatment:
Supportive care
In severe cases, splenectomy has been performed
Stem cell transplants have also been performed
Mitapivat:
Approved by FDA in 2022
Oral small molecule pyruvate kinase activator
Approved based on results of ACTIVATE-T trial:
In 2022, the FDA approved mitapivat, which is an oral, small molecule allosteric activator of red-cell PK
There’s a lot of other enzyme deficiencies that can occur.
TFOC Tip: Most of these deficiencies are autosomal recessive, EXCEPT phosphoglycerate kinase, which is X-linked and adenosine deaminase deficiency which is autosomal dominant

Image source: Table 30.3 from Wintrobe’s Hematology, 14th Edition. No copyright infringement intended.
References:
Wintrobe’s Hematology, 14th Edition
https://ashpublications.org/hematology/article/2005/1/13/19266/Red-Cell-Membrane-Disorders
Perotta et al. Hereditary spherocytosis. Lancet 2008; 372: 1411–26
The crew behind the magic:
Show outline: Ronak Mistry
Production and hosts: Ronak Mistry, Vivek Patel, Dan Hausrath
Editing: Resonate Recordings
Shownotes: Ronak Mistry
Social media management: Ronak Mistry

We are proud to partner with HemOnc.org!
Want to learn more about the trials that lead to the regimens discussed today? What about dosing schedules? See links in the show notes for a link to HemOnc.org