
Episode 110: Sickle Cell Series - Chronic Complications in Sickle Cell Disease
In this week’s episode, we discuss chronic complications that can arise in patients with sickle cell disease. The reality is, that unless we have an outpatient clinic or rotate through an outpatient clinic, there is a small chance that we will come across many of these issues. However, screening for and managing these chronic complications can have significant implications on our patients’ lives.
If you have not done so already, be sure to check out our episode 109 for an introduction to sickle cell disease!
Sickle Cell Disease Chronic Complications (Overview)
SCD can give rise to many acute and chronic clinical complications
Please refer to the graphic below from a 2018 Nature Primer publication for an overview of SCD complications

Image source: https://www.nature.com/articles/nrdp201810 (No copyright infringement intended)
What are the current screening guidelines for patients with sickle cell disease?
In addition to regular history and physical examinations, patients with sickle cell disease should undergo the following at least yearly:
CBC and reticulocyte count
Liver function testing
Creatinine
Serum ferritin (in patients with multiple prior transfusions)
Pulse oximetry
Retinal exam by an ophthalmologist
What is the approach to managing chronic pain?
Chronic pain in SCD patients = pain lasting at least 3 months
Chronic pain patients have altered neural pathways which can exacerbate pain and may be triggered by physical (infection, dehydration) and mental health triggers (screen for anxiety and depression)
PISCES study evaluated pain in SCD patients and found that more than 55% of the time patients felt their pain was poorly managed
Goal of therapy - control pain to optimize function and minimize opioid-related side effects
A multi-modal approach is recommended, including NSAIDs, anticonvulsants, antidepressants, anticonvulsants (e.g. gabapentin), and opioids when needed.
What is “avascular necrosis (AVN)” and how do we manage it?
Sickle cells cause small vessel occlusion, leading to impaired blood flow to bones and avascular necrosis
Most common site = hip (other sites: shoulders, knees, feet, back)
~20% of SCD hospital admissions for pain are related to AVN
Diagnosis
Start with an X-Ray (can show diffuse sclerosis, arthritic changes, femoral head collapse)
For early AVN, obtain an MRI
Management
Conservative (PT, multimodal pain control)
May require joint replacement (poor wound healing a limiting factor)
Why are patients with SCD so prone to develop stroke?
In sub-Sarahan Africa and India, around 50% of children will have an overt or silent cerebral infarct by 18.
The incidence of overt strokes is 24% in HbSS and 10% in HbSC by age 45.
Strokes in SCD adults are a result of several factors:
Low O2 content due to lower oxygen saturation and/or acute drops in hemoglobin
Cerebral vasculopathy compromising blood flow
Cardiac risk factors (as those in the general population) e.g. HTN, DM, HLD, AF, renal disease
Acute infection 2/2 increased metabolic demands
Prior strokes
Rapid increases in hemoglobin, typically >12g/dL, 2/2 to transfusions or autotransfusion from splenic or liver sequestration
Ischemic strokes are more common in patients <20 and >30 years old; whereas hemorrhagic strokes are more common between 20-29 years old.
How do we minimize stroke risk in adults with SCD?
To date, no RCTs to inform primary or secondary stroke prevention in adults
Most data is extrapolated from RCTs in children
In children, a higher transcranial Doppler velocity is associated with a higher risk of stroke
130 patients with mean age of 8.3 years with elevated (>200cm/s) transcranial Doppler measurements randomized to standard of care versus transfusion to threshold hemoglobin S concentration of <30%
Primary endpoint = cerebral infarction & intracranial hemorrhage
92% lower risk of stroke (p<0.001) in transfusion group (1 stroke in transfusion group, 11 strokes in standard-care group)
Follow-up study in 2005 assessing discontinuation of transfusions after 30 months if Doppler measurements normalized
Study stopped early due to high risk doppler findings in 14 and strokes in 2 amongst the 41 children in the transfusion-halted group.
2017 Lancet non-inferiority trial assessing switching transfusions to maximally tolerated hydroxyurea
Children on transfusion therapy after 12 months with normal TCD maintained benefit after transitioning to maximally tolerated hydroxyurea dose
Based on this data, the most recent guidelines from ASH recommend:
In patients with HbSS and HbSB0, annual TCD from ages 2-16
If there is an abnormal TCD: regular transfusions to keep HbS <30%, Hb >9g/dL x1 year
If after 1 year there is a desire to stop transfusions (e.g. due to iron overload), titrate hydrea to max tolerated dose.
How do we work-up and manage SCD patients with pulmonary hypertension (pHTN)
pHTN is a poor prognostic marker in patients with SCD (mortality rate of 36%)
Regular screening for symptoms of pHTN, including SOB, SOB on exertion, excessive fatigue, exertional hypoxia, and chest pain, should be performed
Symptomatic patients should be evaluated for pHTN with an echocardiogram
A tricuspid regurgitant velocity (TRV) greater than or equal to 2.5 m/sec on echo predicts pHTN and increased mortality (though is itself not diagnostic of pHTN)
A right heart catheterization is needed to formally diagnose pHTN
Management:
General pHTN management (vasodilators fluid optimization) under the care of an SCD & pHTN specialist
Aggressive management of underlying SCD
Case series of 5 homozygous SCD adults showing improvement in TRV and hemolytic markers after hydrea at maximally tolerated dose for an average of 14 months
Chronic RBC transfusions can also help, though there is no peer reviewed data to evaluate this.
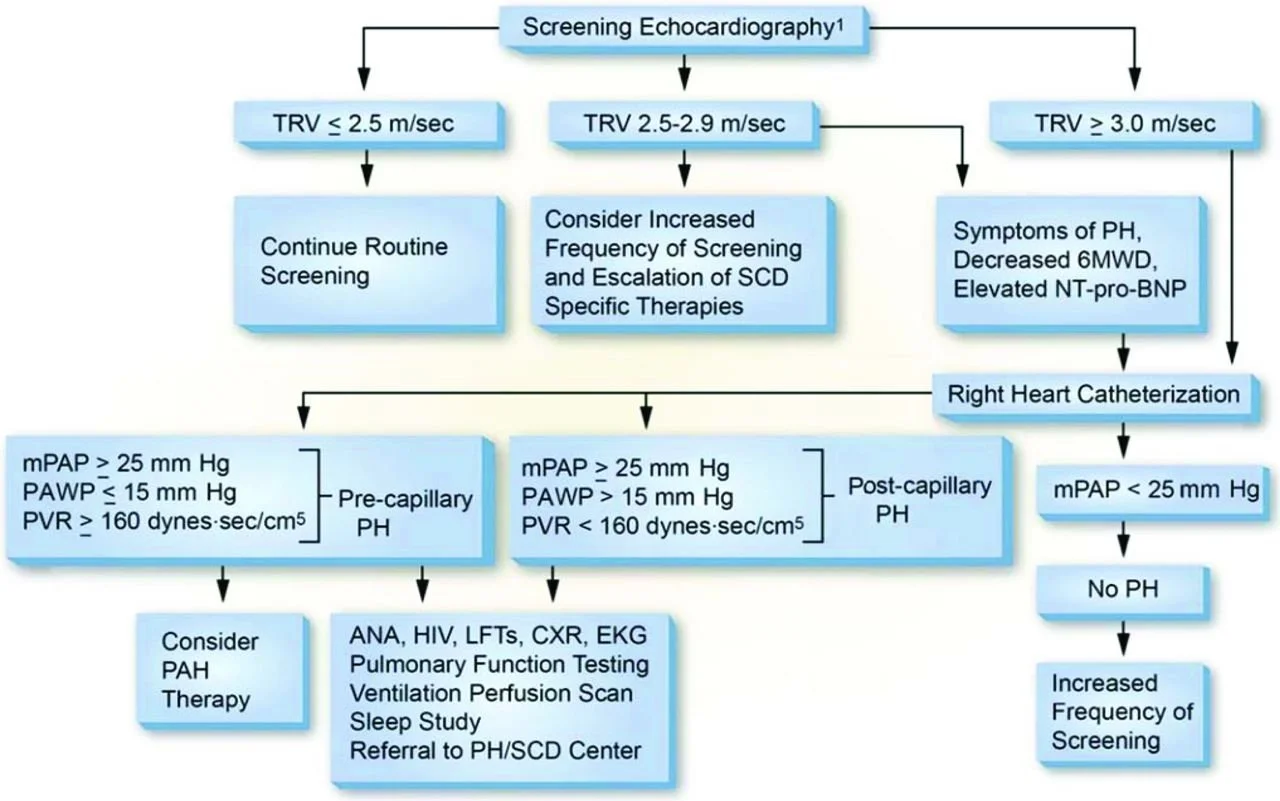
Image source: https://ashpublications.org/hematology/article/2014/1/425/20525/Pulmonary-hypertension-in-sickle-cell-disease (No copyright infringement intended)
How are the kidneys affected by SCD?
Sickle cell nephropathy begins at a young age with glomerular hyperfiltration, progressing to microalbuminuria in late childhood/early adulthood
Most patients do NOT progress any further
A prospective case-control suggests an incidence of 4.2% and 2.4% of advanced renal disease in HbSS patients and in HbSC patients, respectively, with a median age of onset at 23.1 and 49.9 years
A follow up study estimated a rate of 12% in HbSS patients with a median age of onset of 37 years.
A 2008 report identified CKD as the cause of death in 45% of patients who died 60+
HbSS and HbSB0 have higher incidence and degree of microalbuminuria than HbSC or HbSB+
What does sickle cell nephropathy entail?
Hyposthenuria: inability to concentrate urine >450 mOsm/kg under water-deprived conditions
Hyperfiltration: More blood moving through the glomeruli 2/2 anemia and cardiac output (chronic transfusions do not improve this)
Proteinuria: Albumin in urine
Microalbuminuria in 28% of patients 16-25; 38% of patients 26-35; 50% of patients 36-45, and >60% of patients 46+
Nephrotic range proteinuria in 4% of patients
Hematuria
In sickle cell disease and trait
Secondary to renal papillary necrosis
Investigate if progressive, due to the association with medullary cell carcinoma (more common in sickle cell trait)
How should we screen patients and what can we do to help decrease the risk of CKD progression?
Screen with at least once yearly albumin:creatinine ratio and GFR check
Rate of change more important than absolute number (may signal rapidly declining kidney function)
Early referral to nephrology (worse outcomes for ESRD patients not co-managed by nephrology)
How do we manage CKD in SCD patients?
Manage HTN
ACE-inhibitors or ARBs if proteinuria
Hydroxyurea
The BABY HUG study = multicenter study investigating safety and effect of HU on renal function in infants can be safely given to infants and if it influences renal function
HU had no effect on primary outcome, but patients treated with HU had higher urine osmolarities and achieved smaller renal volumes compared to placebo
HUSTLE follow-up study demonstrated that patients 18-23 years old treated with HU for 3 years had better renal function
ESA: Consider for hemoglobin <10mg/dL, as elevated hemoglobin can trigger vaso-occlusive crises
ESRD requiring dialysis carries a very poor prognosis.
One report estimates mean time to death after diagnosis of ESRD requiring HD of 4 years
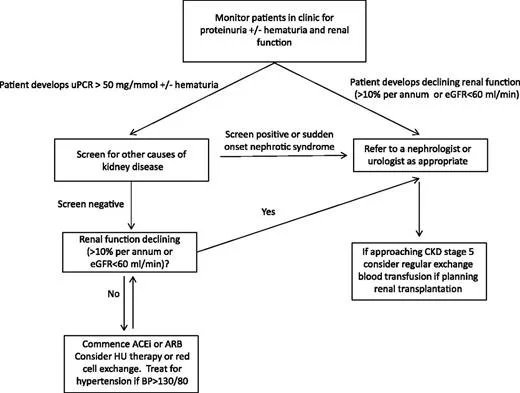
Image source: https://ashpublications.org/blood/article/123/24/3720/32966/How-I-treat-renal-complications-in-sickle-cell (No copyright infringement intended)
How do we manage iron overload in patients with sickle cell disease?
Chronic transfusions increase the risk of iron overload
Increased iron can cause organ damage, including cirrhosis and cardiac dysfunction
One study of 141 patients found that ⅓ of patients had excess iron overload post-mortem; and 7% of deaths were thought to be linked to the effects of iron overload
Check serum ferritin every 3 months in patients who have received multiple transfusions in their lifetime (usually ~20)
Proceed with a liver MRI to measure liver iron content (LIC) if ferritin >1000ng/mL
The units of iron accumulation in the liver is mg Fe/g
If the liver iron content >15mg Fe/g, then recommendations are to consider a cardiac T2* MRI to evaluate for cardiac iron deposition.
Treatment includes iron chelation therapy if LIC >7 mg Fe/g or if ferritin consistently >1000 ng/mL
What are other complications to be aware of?
Leg Ulcers: Due to poor circulation and poor wound healing
Retinopathy: Due to vaso occlusion of retinal arterioles, ischemia and neovascularization, hemorrhage, and autoinfarction. More severe in HbSC than HbSS
Psychosocial complications: missed school and life events, depression, and anxiety
VTE
References:
PISCES Project: McClish, D. K., Penberthy, L. T., Bovbjerg, V. E., Roberts, J. D., Aisiku, I. P., Levenson, J. L., Roseff, S. D., & Smith, W. R. (2005). Health related quality of life in sickle cell patients: The pisces project. Health and Quality of Life Outcomes, 3(1). https://doi.org/10.1186/1477-7525-3-50
STOP Trial: Adams, R. J., McKie, V. C., Hsu, L., Files, B., Vichinsky, E., Pegelow, C., Abboud, M., Gallagher, D., Kutlar, A., Nichols, F. T., Bonds, D. R., Brambilla, D., Woods, G., Olivieri, N., Driscoll, C., Miller, S., Wang, W., Hurlett, A., Scher, C., … Waclawiw, M. (1998c). Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. New England Journal of Medicine, 339(1), 5–11. https://doi.org/10.1056/nejm199807023390102
STOP2 Trial: Adams, R., & Brambilla, D. (2005). Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. New England Journal of Medicine, 353(26), 2769–2778. https://doi.org/10.1056/nejmoa050460
TWITCH Trial: Ware, R. E., Davis, B. R., Schultz, W. H., Brown, R. C., Aygun, B., Sarnaik, S., Odame, I., Fuh, B., George, A., Owen, W., Luchtman-Jones, L., Rogers, Z. R., Hilliard, L., Gauger, C., Piccone, C., Lee, M. T., Kwiatkowski, J. L., Jackson, S., Miller, S. T., … Adams, R. J. (2016). Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia—TCD with transfusions changing to hydroxyurea (twitch): A Multicentre, open-label, phase 3, non-inferiority trial. The Lancet, 387(10019), 661–670.
DeBaun, M. R., Jordan, L. C., King, A. A., Schatz, J., Vichinsky, E., Fox, C. K., McKinstry, R. C., Telfer, P., Kraut, M. A., Daraz, L., Kirkham, F. J., & Murad, M. H. (2020). American Society of Hematology 2020 Guidelines for Sickle Cell Disease: Prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Advances, 4(8), 1554–1588. https://doi.org/10.1182/bloodadvances.2019001142
Platt, O. S., Brambilla, D. J., Rosse, W. F., Milner, P. F., Castro, O., Steinberg, M. H., & Klug, P. P. (1994). Mortality in sickle cell disease -- life expectancy and risk factors for early death. New England Journal of Medicine, 330(23), 1639–1644. https://doi.org/10.1056/nejm199406093302303
Serjeant, G. R., Serjeant, B. E., Mason, K. P., Hambleton, I. R., Fisher, C., & Higgs, D. R. (2009). The changing face of homozygous sickle cell disease: 102 patients over 60 years. International Journal of Laboratory Hematology, 31(6), 585–596. https://doi.org/10.1111/j.1751-553x.2008.01089.x
Day, T. G., Drasar, E. R., Fulford, T., Sharpe, C. C., & Thein, S. L. (2011). Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica, 97(2), 201–205. https://doi.org/10.3324/haematol.2011.050336
Bakir, A. A., Hathiwala, S. C., Ainis, H., Hryhorczuk, D. O., Rhee, H. L., Levy, P. S., & Dunea, G. (1987). Prognosis of the nephrotic syndrome in sickle glomerulopathy. American Journal of Nephrology, 7(2), 110–115. https://doi.org/10.1159/000167444
McClellan, A. C., Luthi, J., Lynch, J. R., Soucie, J. M., Kulkarni, R., Guasch, A., Huff, E. D., Gilbertson, D., McClellan, W. M., & DeBaun, M. R. (2012). High one year mortality in adults with sickle cell disease and end‐stage renal disease. British Journal of Haematology, 159(3), 360–367. https://doi.org/10.1111/bjh.12024
Darbari, D. S., Kple-Faget, P., Kwagyan, J., Rana, S., Gordeuk, V. R., & Castro, O. (2006). Circumstances of death in adult sickle cell disease patients. American Journal of Hematology, 81(11), 858–863. https://doi.org/10.1002/ajh.20685
Porter, J., & Garbowski, M. (2013). Consequences and management of iron overload in sickle cell disease. Hematology, 2013(1), 447–456. https://doi.org/10.1182/asheducation-2013.1.447
Treadwell, M. J. (2023). Mental health and psychological resilience in sickle cell disease. The Lancet Haematology, 10(8). https://doi.org/10.1016/s2352-3026(23)00166-7
The crew behind the magic:
Show outline: Ronak Mistry
Production and hosts: Ronak Mistry, Vivek Patel, Dan Hausrath
Editing: Resonate Recordings
Shownotes: Karam Elsolh
Social media management: Ronak Mistry

We are proud to partner with HemOnc.org!
Want to learn more about the trials that lead to the regimens discussed today? What about dosing schedules? See links in the show notes for a link to HemOnc.org
Have some extra time and want to make some extra money? Click here to get paid to participate in market research surveys!