
Episode 109: Sickle Cell Series - Introduction to Sickle Cell Disease
It’s time for another new series, this time focusing on Sickle Cell Disease! In this first episode, we lay the foundation for our future discussions. This episode can be dense, but rest assured we will revisit these concepts and expand on them throughout the series.
What is hemoglobin?
Adult hemoglobin, also called Hemoglobin A or Hemoglobin A1, is a metalloprotein comprising two beta-globin chains and two alpha-globin chains.
Two beta-globin alleles (the HBB gene) located on chromosome 11
Four alpha-globin alleles (the HBA gene) located on chromosome 16
There are additionally a number of different globin genes similar to beta globin on chromosome 11:
HBE: involved in embryonic hemoglobin
HBG1 and HBG2: which make fetal hemoglobin
HBD: which makes a variant hemoglobin A
The protein component of hemoglobin is bound to four heme groups, each of which has an iron atom at the center.
The iron atom holds onto oxygen molecules, and the whole enzyme changes conformation when oxygen binds. This leads to a change in how tightly hemoglobin binds to oxygen with each successive oxygen molecule that gets attached, resulting in the “S-curve” of oxygen affinity.
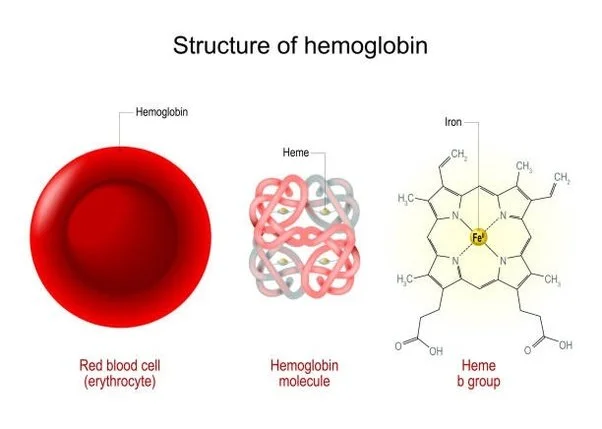
Image source: https://www.quora.com/How-does-the-structure-of-haemoglobin-enable-it-to-carry-out-its-functions (No copyright infringement intended)
What is the issue in sickle cell disease?
Sickle cell disease results when there is an abnormal variant in the beta globin chain: valine for glutamate at the 7th amino acid (note that this was previously referred to as the 6th position - papers prior to the early 2000s will refer to it as such).
The abnormal hemoglobin molecule can polymerize when in its de-oxygenated state forming a long crystal that deforms the red cell into the classic “sickle” shape

Image source: https://www.nationwidechildrens.org/conditions/sickle-cell-disease (No copyright infringement intended)
Why are sickled cells problematic?
Sickled red cells are highly pro-inflammatory, adhere to the endothelium, and can spontaneously break down
Intravascular hemolysis causes diffuse endothelial dysfunction and vasospasm from free heme scavenging nitric oxide and is highly prothrombotic from exposed phosphatidylserine on the shards of RBC membrane
The sickle cells obstruct blood flow in the microvasculature, resulting in a positive feedback cycle can occur where progressive vasoocclusion causes increased tissue hypoxia, which causes further vasoocclusion
Chronic hemolysis and low-level vasooclusion causes other issues as well including chronic anemia, typically in the 6-10 range for symptomatic sickle cell disease patients, functional asplenia, and chronic kidney disease.
What is the difference between sickle cell disease and sickle cell trait?
Patients who are heterozygous for the abnormal beta chain are said to have sickle cell trait
Sickle cell trait confers protection against malaria, therefore the epidemiology mirrors that of where malaria is most prevalent, including sub-saharan Africa, India, Greece, and the Middle East.
Clinically, how does sickle cell trait differ from sickle cell disease?
If the non-sickle beta chain is normal, most heterozygotes will not have any symptoms, and will have a normal baseline hemoglobin concentration.
There is, however, an increased risk of kidney issues in patients with sickle cell trait:
Hematuria from small renal infarcts; some studies estimating 50% increased risk of CKD compared to the general population
Renal medullary carcinoma is also seen in sickle cell trait; appropriate workup for patients with sickle cell trait with hematuria is important.
What are other genotypic variations that can present like sickle cell disease/HbSS?
The symptoms classically associated with sickle cell disease emerge when there is no normal beta globin chain around to dilute the sickle beta globin. Most commonly, this is in patients with homozygous sickle cell mutations. Patients with Sickle Cell Disease have genotype HbSS
Patients can also be “compound heterozygotes” meaning they have one HbS gene and another variant beta globin gene, with the most common being:
Hemoglobin C: Broadly found in the West African population (17-28% of individuals in West Africa, referred to as HbSC genotype
Beta thalassemia variants, beta+ and beta0 thalassemia, referred to as HbSbeta+ and HbSbeta0 genotype
HbOarab: This variant can be as bad or worse than HbSS in terms of rate of vaso-occlusive complications
HbSC disease and HbSbeta+ genotypes produce milder phenotypes than HbSS with less frequent and less severe complications, but patients can still have any of the same complications as those with SS
HbSbeta0 produces a phenotype that is nearly identical to HbSS, given that in both situations the only beta globin chain the person produces is the sickle beta globin.
Penn State, has an extensive database of the many other beta globin variants that you might encounter: http://globin.cse.psu.edu/hbvar
How do you determine a patient’s genotype?
Hemoglobin electrophoresis is still the current standard for diagnosis, but with the increasing availability and speed of DNA sequencing, we will probably be increasingly going towards genetic testing to make our diagnoses.
The electrophoresis method (or related methods isoelectric focusing and HPLC) use biochemical techniques to separate different hemoglobin variants and measure their proportions
Fun fact: Nobel Laureate Linus Pauling discovered in 1949 that patients with sickle cell disease have a pattern different than normal hemoglobin on electrophoresis.
Normal breakdown:
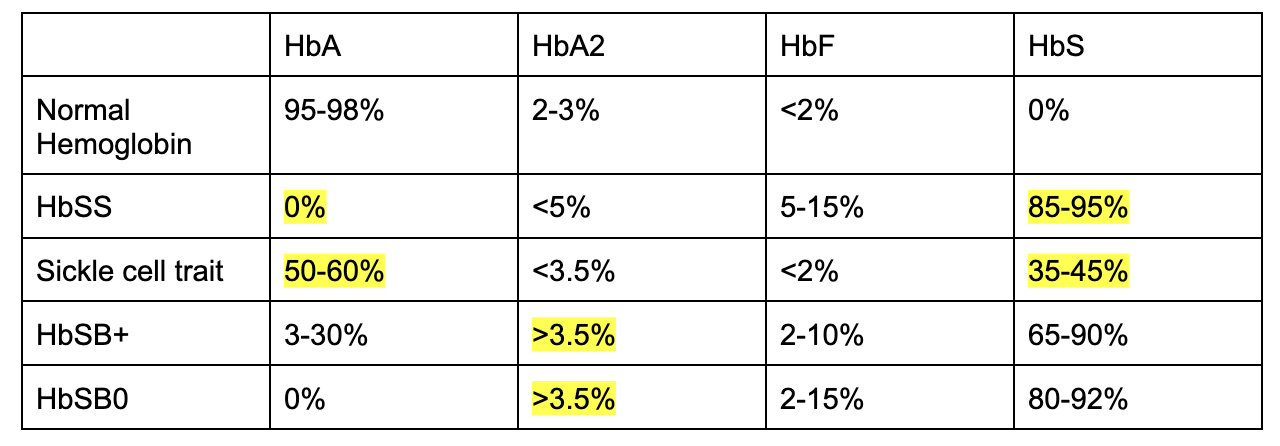
Note key differences highlighted in yellow!
What is “hereditary persistence of fetal hemoglobin”?
Patients with this have naturally increased level of fetal hemoglobin which helps counteract some of the detrimental effects of hemoblobin S, and they will have fewer vaso-occlusive complications
References:
ALLISON AC. POLYMORPHISM AND NATURAL SELECTION IN HUMAN POPULATIONS. Cold Spring Harb Symp Quant Biol. 1964;29:137-49. doi: 10.1101/sqb.1964.029.01.018. PMID: 14278460.
Eaton WA, Bunn HF. Treating sickle cell disease by targeting HbS polymerization. Blood. 2017 May 18;129(20):2719-2726. doi: 10.1182/blood-2017-02-765891. Epub 2017 Apr 6. PMID: 28385699; PMCID: PMC5437829.
Gowda L, Vege S, Kessler D, Shaz B, Westhoff CM. Screening of blood donors for sickle cell trait using a DNA-based approach: Frequency in a multiethnic donor population. Transfusion. 2021 Jul;61(7):2008-2013. doi: 10.1111/trf.16403. Epub 2021 Apr 30. PMID: 33929058.
PAULING L, ITANO HA, et al. Sickle cell anemia a molecular disease. Science. 1949 Nov 25;110(2865):543-8. doi: 10.1126/science.110.2865.543. PMID: 15395398.
Sheehan VA, Gordeuk VR, Kutlar A. Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities. In: Kaushansky K, Prchal JT, Burns LJ, Lichtman MA, Levi M, Linch DC. eds. Williams Hematology, 10e. McGraw-Hill Education; 2021. Accessed July 16, 2024.
The crew behind the magic:
Show outline: Daniel Hausrath
Production and hosts: Ronak Mistry, Vivek Patel, Dan Hausrath
Editing: Resonate Recordings
Shownotes: Ronak Mistry
Social media management: Ronak Mistry

We are proud to partner with HemOnc.org!
Want to learn more about the trials that lead to the regimens discussed today? What about dosing schedules? See links in the show notes for a link to HemOnc.org
Have some extra time and want to make some extra money? Click here to get paid to participate in market research surveys!